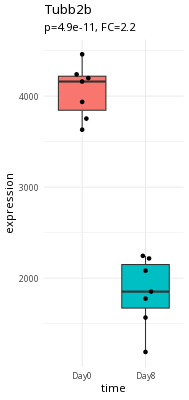
Here is an example of a large expression difference (Tubb2a).
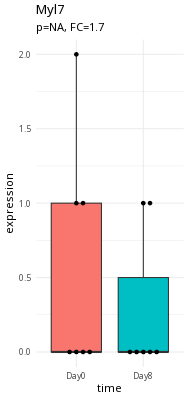
Here is an example of a questionable expression difference (myl7).
Take a second and think about what makes Tubb2a a better candidate than myl7.
3.3 What are we trying to accomplish?
In RNAseq, we are interested in assessing expression differences between samples at either the gene or transcript level.
In order to do this, we need phenotype information about the samples in our experiment. Specifically, we need to have information about:
conditions (infection, time)
possible confounders (gender)
3.4 Experimental Design of RNAseq Experiments
When we design our RNAseq experiment, we want to be able to:
Number and type of replicates
Avoiding confounding
Addressing batch effects
The important thing to note is that if you’re unsure about the experimental design, contact the Bioinformatics Core. They can help you design the experiment based on what samples you have.
We’ll talk much more about experimental design next week.
3.5 What is Bioconductor?
Data in bioinformatics is often complex. To deal with this, developers define specialised data containers (termed classes) that match the properties of the data they need to handle.
This aspect is central to the Bioconductor1 project which uses the same core data infrastructure across packages. This certainly contributed to Bioconductor’s success. Bioconductor package developers are advised to make use of existing infrastructure to provide coherence, interoperability, and stability to the project as a whole.
3.6 S4 Bioconductor Objects
At the heart of Bioconductor are the S4 Objects. You can think of these data structures as ways to package together:
Assay Data, such as an RNAseq count matrix
Metadata (data about the experiment), including Experimental Design
Part of why Bioconductor works is this data packaging. We can write functions to act on the data in these S4 Objects as part of a processing workflow. As long as we output a Bioconductor S4 object, our routines can work as part of a pipeline. These routines are called methods(), and may come from a variety of packages.
NoneWhat is a method?
You may have heard of functions and methods - what is the difference?
A working definition of a method is that it is a function that works on a particular object type, such as the SummarizedExperiment type we’re going to investigate in a little bit.
Method is a little bit more specific than a function.
You can think of the Bioconductor S4 objects as taking the place of data.frames in dplyr pipelines - they are the common format that all of the Bioconductor methods work on. They allow Bioconductor methods to be interoperable - we can mix and match methods from various packages to customize our analysis.
3.6.1 Validation
The other big part of S4 objects is validation. This can be a bit hard to wrap your head around.
The Bioconductor designers put special validation checks on the input data for the Bioconductor objects when you load data into them. The following is what is called the Constructor for the SummarizedExperiment object. This is what we use to make a brand new SummarizedExperiment object from its pieces.
Each argument to the SummarizedExperiment constructor defines restrictions on that slot. For example, there is a slot called assays.
The checkDimnames argument is critical. In order to do any work with an experiment you need to map samples to colNames (the experimental matrix). For example, to calculate differential expression between samples, you need to specify the different groups to compare and which samples map to which groups. Thus, the column names in the AssayData must be identical to the row names in colData.
NoneIf it isn’t in colData, it doesn’t exist
Keep in mind that your colData must be as complete as possible. Why? The short answer is that if there isn’t a row for your sample in colData, then it basically doesn’t exist for Bioconductor methods.
So make sure your colData contains sample names for all your samples.
3.7SummarizedExperiment
The following diagram shows how the different data slots in the SummarizedExperiment relate to each other.
We’ll take a look at data in a SummarizedExperiment object:
In some ways, the SummarizedExperiment object is like a data.frame, but with extra metadata. For example, our object has column names, which correspond to sample identifiers:
Assay Data is very flexible. For example, there are flow cytometry objects where the rows correspond to cell surface markers, and columns that correspond to each cell. Similarly, SingleCellExperiment objects (which are derived from SummarizedExperiment) have rows that correspond to Genes and columns that correspond to individual cells.
3.8.1 Think about it
Are the colnames of our object identical to the colnames of the assay object? Try it out:
Metadata is information about the experiment that is not part of the Assay Data.
The most important part of the metadata is the colData slot. This slot contains information about the samples (the columns) of the assay. This is where we store the Experimental Design that we talked about.
It might be tempting to extract the assay data and the metadata and work with them separately. But as we’ll see in the following section, these two slots work together and enable all sorts of analysis.
3.10 Subsetting SummarizedExperiment
We saw that we have the checkNames constraint. This is because we can use the colData and the assayData in our object to do subsetting using the metadata.
This tight interaction of metadata and assay data is critical when we start doing differential analysis. The experimental design will help determine whether we can make the comparisons we want to make and the conclusions we can draw from the dataset.
NoneRemember the Comma
Remember that the , (comma) is used to distinguish between rows and columns. Rows correspond to genes and columns correpsond to samples.
In the above example, we are subsetting the samples, so our criteria goes after the comma.
For manipulating the SummarizedExperiment object, we will use the tidySummarizedExperiment package to subset it - essentially this package lets us use dplyr on the SummarizedExperiment and DESeqDataset objects.
3.10.1 Try it out
Try subsetting GSE96870 to have time == "Day0". How many samples are left?
The SummarizedExperiment object does not act like the normal data.frame/tibble we expect, especially in filtering and subsetting.
The tidySummarizedExperiment package will be helpful for us to understand and visualize the SummarizedExperiment package. Once we do that, our SummarizedExperiment object will act more like a data.frame.
Basically, tidySummarizedExperiment treats the data as a long data frame that combines both the assay and metadata. This format is helpful in doing more work with the tidyverse:
The following object is masked from 'package:generics':
tidy
GSE96870
Warning: `when()` was deprecated in purrr 1.0.0.
ℹ Please use `if` instead.
ℹ The deprecated feature was likely used in the tidySummarizedExperiment
package.
Please report the issue at
<https://github.com/stemangiola/tidySummarizedExperiment/issues>.
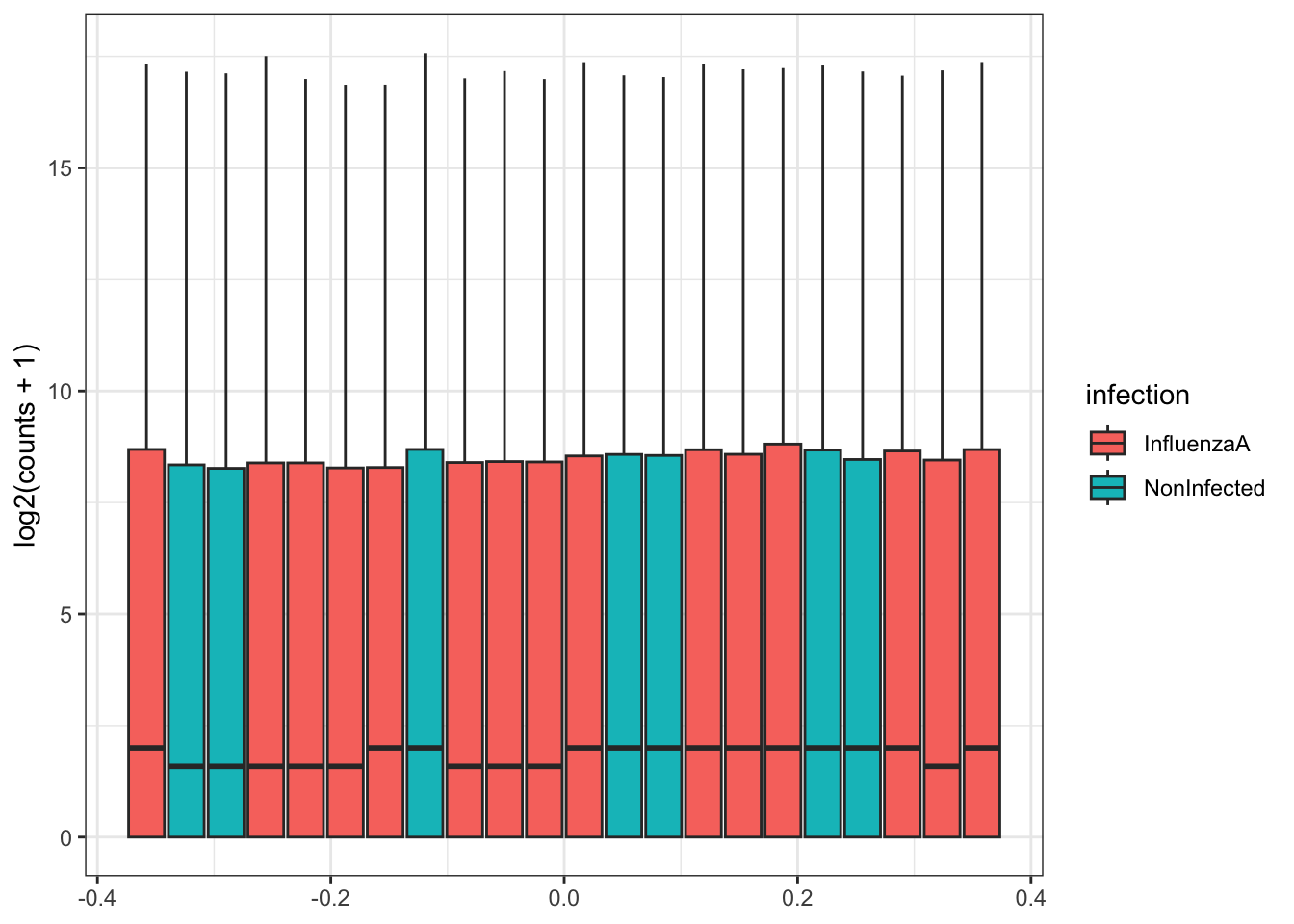
Our data (individual gene counts) is highly skewed. If we log-transform the data, then the skew is lessened. The boxplots of log counts are helpful for detecting batch effects.
Take a look at the above box plot. Are there differences in distribution between samples?
In order to compare expression levels between samples, we will need to adjust, or normalize by library size. We’ll investigate this further when we get to differential expression.
We package multiple parts of the data (assay data and metadata) into a SummarizedExperiment object:
Columns correspond to samples, Rows correspond to gene locus or transcripts
Column names of the assay data must match a column in the colData
Experimental Design variables are part of the colData
Rownames of the assay data must match the rowNames in the column data
The SummarizedExperiment object lets us subset by phenotype variables
tidySummarizedExperiment lets us interact with the SummarizedExperiment object as if it were a long data frame with both metadata and assay data together.
We can plot and subset the SummarizedExperiment object using our regular tidyverse tools (ggplot2, dplyr, etc.) once we load the tidySummarizedExperiment package.
NoteWhat about all of the other kinds of objects?
Most of the other Bioconductor Data Structures derive from some variant of the SummarizedExperiment. They might add some functionality that is core to the package they belong to, such as DEseq2. This includes seurat objects for Single Cell sequencing.
The Bioconductor project was initiated by Robert Gentleman, one of the two creators of the R language. Bioconductor provides tools dedicated to omics data analysis. Bioconductor uses the R statistical programming language and is open source and open development.↩︎